

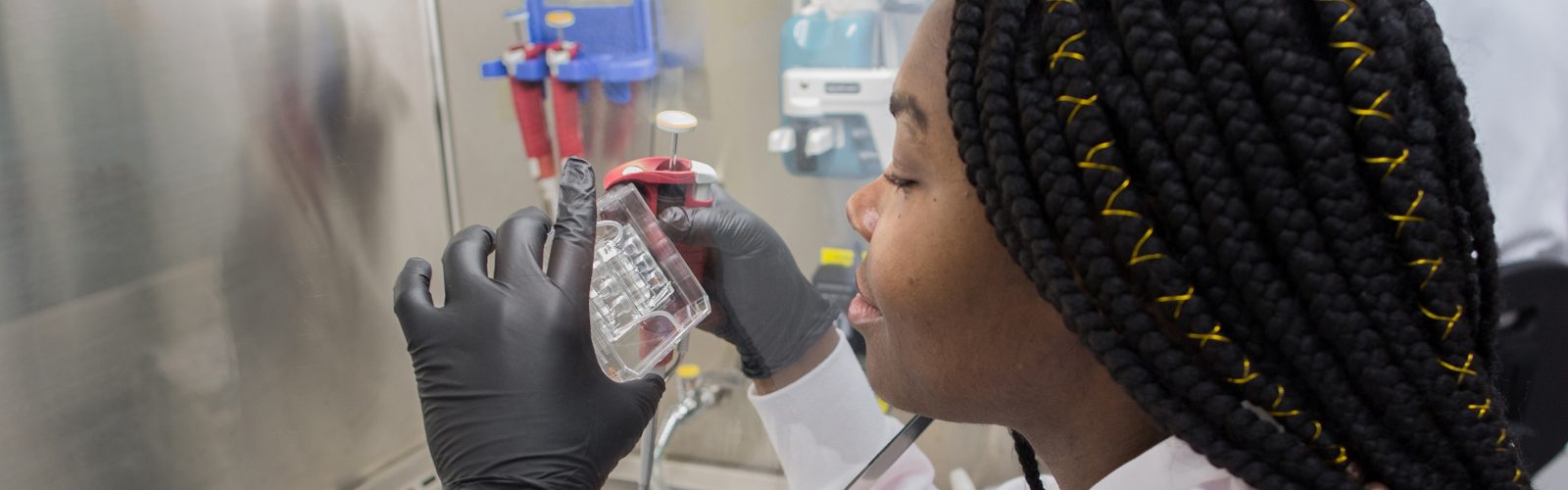









We strive to create an inclusive, collaborative and connected culture that fosters cutting-edge basic, translational, education, and health outcomes research. Our research programs investigate the underlying mechanisms of disease, translate discoveries in the laboratory to clinical trials, and offer experimental options with the goal of improving patient care. Our research funding portfolio exceeds $18 million annually with a large percentage from the National Institutes of Health.
Our Researchers & Labs >>
Latest Research News
A Rare Cancer, A Relentless Advocate
Anal cancer is a rare cancer in humans, but its incidence and mortality has steadily increased over the last decade in the U.S. Despite that growth, stigma surrounding anal cancer increasingly stifles the conversation around …
Dr. Evie Carchman Awarded Distinguished Faculty Postdoc Mentoring Award
Dr. Evie Carchman, associate professor in the Division of Colorectal Surgery, was awarded the Distinguished Faculty Postdoc Mentoring Award by the UW–Madison Postdoctoral Association at the annual Celebration of Postdoc Excellence on May 22. The …
Dr. Nabeel Zafar Awarded Dean’s Award for Excellence in Medical Student Research Mentorship
Dr. Nabeel Zafar, assistant professor in the Division of Surgical Oncology, was awarded the Dean’s Award for Excellence in Medical Student Research Mentorship at the UW School of Medicine and Public Health’s Medical Education Day …
Dr. Sam Poore to lead clinical trials education program
The project, Partnership to Expand Education and Training Pathways in Clinical Research is led by Samuel Poore, MD, PhD, professor, Department of Surgery, UW School of Medicine and Public Health; and co-investigators Rafael Veintimilla, MD, senior director, …
Against the Odds: Advancing Research for an Ultra-Rare Liver Cancer
When talking about cancer research, much of the conversation centers around breast, prostate, lung and colorectal cancer – and rightly so as those cancers account for nearly 50% of all new cancer diagnosis in the …
Wisconsin Surgery Research Roundup: March 2026
Wisconsin Department of Surgery members engage in remarkable research that yields many impactful publications every month. We’re highlighting several of these publications monthly to showcase the diversity of research in the department; see selections from …
Wisconsin Surgery Research Roundup: February 2026
Wisconsin Department of Surgery members engage in remarkable research that yields many impactful publications every month. We’re highlighting several of these publications monthly to showcase the diversity of research in the department; see selections from …
Dr. Patrick Carney Wins Flash Talk at UW Carbone Cancer Center Research Retreat
Dr. Patrick Carney, general surgery resident, won the flash talk session at the UW Carbone Cancer Center’s Annual Research Retreat. The top six abstracts from the meeting were selected for a 10-minute oral presentation and …
Dr. Ana De Roo Awarded Prestigious Career Development Award from the American Cancer Society
Division of Colorectal Surgery Assistant Professor Ana De Roo, MD, MSc hopes to spend her career improving clinical processes and decision-making so patients with cancer are supported, informed, and receive care that is aligned with …
Opportunities for Success: Residents Thrive During Research Years
Research is one of the foundational pillars in the UW–Madison Department of Surgery. That is reflected throughout the department, including the trainee programs. All three of our residency programs provide trainees opportunities for dedicated research …
Wisconsin Surgery Research Roundup: January 2026
Wisconsin Department of Surgery members engage in remarkable research that yields many impactful publications every month. We’re highlighting several of these publications monthly to showcase the diversity of research in the department; see selections from …
Department of Surgery Celebrates 17th Annual Research Summit
On Thursday, Jan. 29, the Department of Surgery celebrated the 17th Annual Research Summit. The theme of this year’s summit was “Research Without Borders.” Our keynote speaker was Dr. Sandra Wong, Dean of Emory University …
General Surgery Resident Publishes Research about Burnout
Dr. Sydney Tan, general surgery resident, recently had her research, “Work Hours, Stress, and Burnout Among Resident Physicians,” published in JAMA Network Open. Dr. Tan and her research partners at the University of Wisconsin–Madison examined …
Odorico Lab Awarded Fall Research Competition Grant to Study How Engineered Stem Cell-Derived Islets Evade the Immune System
Over the last five years there have been significant advancements in the treatment of Type 1 diabetes, including the 2023 Food and Drug Administration approval of the first stem cell-derived therapy that can replace the …
Dr. Anna Beck Awarded Career Development Grant from UW BIRCWH Scholars Program
Congratulations are in order for Division of Surgical Oncology Assistant Professor Anna Beck, MD, who was recently awarded a position in UW’s 2025-2027 cohort of the Building Interdisciplinary Research Careers in Women’s Health (BIRCWH) scholars …
- More Research posts

Research Leadership

Luke Funk, MD, MPH
Vice Chair of Research

Angela Gibson, MD, PhD
Vice Chair of Research

JoAnne Vaccaro
Director of Research Operations
Office of Clinical Research
Provides coordinator and regulatory support for all types of human subjects research including sponsored clinical trials, survery studies, registries, and chart reviews.
UW Humanized Mouse Core
The Humanized Mouse Core at the University of Wisconsin–Madison provides investigators with a variety of humanized mouse models for their individual research needs.
Statistical Analysis and Research Programming (STARP) Core
Provides faculty, researchers, and residents/fellows/students with comprehensive statistical and research programming support for their research activities including study design, grant application, abstract/manuscript review, statistical analysis, programming and results interpretation.
WiSOR Qualitative Core
The WiSOR Qualitative Core offers faculty and staff consultation, support, and expertise on study design, methodology, data collection and analysis, and interpretation of results. It also offers transcription services.